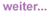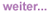CHRISTIAN HOFFMANN, HAMBURG
Opportunistische Infektionen - Teil 6: Multizentrischer Morbus Castleman
Obgleich im eigentlichen Sinn weder eine echte opportunistische Infektion noch eine klassische AIDS-Erkrankung, ist der HIV-assoziierte Multizentrische Morbus Castleman (MCD) auch heute noch eine höchst problematische Krankheit. Sie stellt die Behandler vor große diagnostische und therapeutische Herausforderungen. Die meist in Schüben schwer kranken Patienten durchleben nicht selten über lange Monate und manchmal sogar Jahre hinweg diagnostische Irrwege, und zwar auch, weil oft weder Kliniker noch Pathologen diese Entität kennen.
Im Gegensatz zum klassischen Morbus Castleman, einer gewöhnlich benignen und lokalisierten Hyperplasie des lymphatischen Gewebes, die von dem US-Pathologen Benjamin Castleman im Jahre 1956 erstmals beschrieben wurde, ist der HIV-assoziierte Multizentrische Morbus Castleman (HIV-MCD) eine lymphoproliferative Erkrankung mit malignem Charakter. Obwohl weder AIDS-definierend noch tatsächlich ein malignes Lymphom, hat der HIV-MCD eine eher ungünstige Prognose. In der prä-HAART-Ära lag die mediane Überlebenszeit nach Diagnose bei einigen Monaten bis wenigen Jahren, in HAART-Ära beträgt die Mortalität immerhin noch bis zu 30%.
Die Pathogenese des HIV-MCD bleibt nur unvollständig verstanden. Entscheidend ist wohl eine aktive Koinfektion mit dem Humanen Herpes-Virus 8 (HHV-8), weshalb es nicht verwundert, dass etwa die Hälfte der Patienten zusätzlich an einem Kaposi-Sarkom leidet. HHV-8 ist in der Lage, die Produktion eines viralen Interleukins zu induzieren, das dem humanen Interleukin-6 sehr ähnlich ist ("vIL-6"). Diese Dysregulation des Zytokinmusters durch die viral produzierten Interleukine scheint entscheidend zur Klinik beizutragen. Vor allem Interleukin-6 und Interleukin-10 sind in enger Assoziation zur HHV-8-Viruslast erhöht. Das Ausmaß der Immunschwäche bzw. CD4-Zelldepletion scheint dagegen pathogenetisch nur eine untergeordnete Rolle zu spielen. Der HIV-MCD kann also durchaus auch bei Patienten mit noch normalem Immunstatus und niedriger HI-Viruslast auftreten. So lag die Viruslast einer neueren Arbeit zufolge zum Zeitpunkt der HIV-MCD-Diagnose bei 27/43 Patienten unterhalb von 400 Kopien/ml. Eine "Entartung" zu malignen Non-Hodgkin-Lymphomen (NHL) ist häufig. Von 60 HIV-MCD-Patienten entwickelten 14 nach einer medianen Beobachtungszeit von 20 Monaten ein malignes Lymphom. Typischerweise werden dabei HHV-8-assoziierte, sonst auch bei HIV-Patienten eher seltene NHL-Subtypen beobachtet, darunter zum Beispiel plasmablastische Lymphome oder auch so genannte Primary Effusion-Lymphome.
KLINIK
Im Vordergrund stehen die meist sehr eindrucksvollen Lymphknotenschwellungen, die palpatorisch weich (wie bei Tuberkulose) bis steinhart (wie bei Lymphom) sein können. Sie treten oft ubiquitär und selten einseitig auf. Bestimmte Prädilektionsstellen gibt es nicht. Hinzu kommt fast immer eine erhebliche B-Symptomatik mit Fieber, Nachtschweiß und Gewichtsabnahme. Fast alle Patienten berichten über Schwäche und ein schweres Krankheitsgefühl. Die Milz ist immer deutlich bis massiv vergrößert. Eine Hepatomegalie (70%), respiratorische Symptome (65%), Ödemneigung bei Hypalbuminämie (55%) sind ebenfalls in der Mehrzahl der Fälle zu finden.


Abb. 1a und b: Histologische Befunde bei HIV-MCD. Keimzentrum mit typischer, zwiebelschalenartiger Schichtung des
Follikelmantels (Abb. a, Giemsa). In Abbildung b (CD21-Färbung) typische Vergröberung des FDC-Netzwerkes
bei regressiv verändertem Keimzentrum
Das Ausmaß der Symptome ist allerdings sehr variabel und fluktuiert mitunter erstaunlich. Typischerweise verläuft die Erkrankung in Schüben, die einige Tage bis Wochen anhalten und in denen die Patienten oft hoch fiebern und schwerkrank sind. Die Schübe werden von längeren, meist mehrwöchigen Perioden unterbrochen, in denen es den Patienten wieder relativ gut geht. Ohne jede Maßnahme können sich dabei auch sehr ausgeprägte Lymphknoten-Schwellungen wieder vorübergehend zurückbilden, so dass sie nicht mehr palpabel sind. Mit zunehmender Dauer der Erkrankung nimmt die Frequenz der Schübe in der Regel zu.
DIAGNOSTIK
Die Diagnose wird histologisch aus einem exstirpierten Lymphknoten gestellt. Voraussetzung ist allerdings, dass der Pathologe auch weiß, wie ein HIV-assoziierter multizentrischer Morbus Castleman histologisch aussieht. Die Keimzentren der Lymphknoten erscheinen zwiebelschalenartig geschichtet (Abb. 1). Man unterscheidet histologisch einen hyalin-vaskulären und einen plasmazellreichen Typ des Morbus Castleman. Letzterer ist typisch für den HIV-MCD. Kliniker sollten den Pathologen explizit auf den Verdacht eines HIV-MCD hinweisen. Bei typischer Symptomatik, d.h. schubweiser Verlauf mit B-Symptomatik, Splenomegalie, hohes C-reaktives Protein (CRP) und fluktuierende Lymphknotenschwellungen, man sich als Kliniker mit der Diagnose einer HIV-assoziierten Lymphadenopathie auf keinen Fall zufrieden geben. HIV alleine macht niemals so krank wie ein MCD. Möglicherweise werden viele Fälle nie korrekt diagnostiziert.
Sonografisch zeigt sich eine vergrößerte Leber und eine meist eindrucksvolle Splenomegalie. Im Labor fallen ein während der Schübe obligat erhöhtes CRP, eine Hypergammaglobulinämie sowie eine Hypalbuminämie ins Auge. Einen MCD ohne ein zumindest passager erhöhtes CRP gibt es nicht. Oft besteht zudem eine deutliche Anämie (mitunter hämolytisch, oft im Rahmen einer Panzytopenie). Fast immer ist die HHV-8-Serologie positiv.
Nach unserer Erfahrung eignet sich vor allem das Akutphase-Protein CRP als diagnostischer Verlaufsparameter, um neben der Klinik den Erfolg zu überwachen. Während eines Schubs sind Werte von weit mehr als 100 mg/l die Regel, zwischen den Schüben finden sich dagegen oft Normalwerte. Mitunter geht die CRP-Erhöhung den klinischen Beschwerden etwas voraus. Das CRP sollte bei Patienten mit MCD deshalb bei jeder Blutentnahme mitbestimmt werden. In der Tabelle 1 sind die wichtigsten Symptome und klinischen Merkmale des HIV-MCD aufgeführt.
THERAPIE

Abb. 2: Hepatosplenomegalie bei MCD


Bei einem HIV-assoziierten MCD muss zügig gehandelt werden. Der Verlauf kann höchst fulminant, die hochfieberhaften Schübe durchaus letal sein. Allerdings fehlen randomisierte Studien und Therapieempfehlungen. Überdies gibt es bislang kein einziges Medikament, das für den MCD offiziell zugelassen ist. Eine antiretrovirale Therapie sollte möglichst zusätzlich gegeben werden, obgleich sie leider am Verlauf bzw. an der Klinik nur wenig ändert. Es sind sogar Fälle beschrieben worden, die erst unter ART auftraten oder sich verschlimmerten, was zu der Vermutung führte, dass die inflammatorische Komponente des MCD durch die Immunrekonstitution sogar verstärkt wird.
Rituximab:
Angesichts eigener Erfahrungen und der neueren Datenlage ist inzwischen der monoklonale Antikörper Rituximab die Therapie der Wahl beim HIV-MCD. Der Wirkmechanismus dieses Antikörpers, der sich gegen CD20-exprimierende Zellen richtet und der für die Behandlung von B-Zell-Lymphomen entwickelt wurde, ist beim HIV-MCD nicht definitiv geklärt. Es ist jedoch wahrscheinlich, dass Rituximab einen Großteil der B-Zellen eliminiert, die von HHV-8 vorwiegend in der Mantelzone des Lymphknotens infiziert werden. Dadurch wird der Pool der Interleukin-6-produzierenden Zellen reduziert. In 2007 wurden zwei größere Studien mit sehr ermutigenden Resultaten publiziert. In einer französischen Studie erreichten immerhin 16/24 Patienten durch vier Kurse Rituximab eine komplette Remission nach einem Jahr. Das Gesamtüberleben lag nach einem Jahr bei 92%, das krankheitsfreie Überleben immerhin bei 74%. In einer englischen Kohorte erreichten 20/21 eine klinische Remission, 14/21 eine radiologische Response. Das Gesamtüberleben lag in dieser Studie nach zwei Jahren sogar bei 95%, das krankheitsfreie Überleben bei 79%. Sowohl CRP als auch Immunglobuline und HHV-8-Viruslast sanken unter Rituximab. Gegeben wird Rituximab in der Dosis von 375 mg pro m2 Körperoberfläche, einmal wöchentlich über jeweils 4 Wochen. Auf eine sehr gute Hydratation ist zu achten. Nach unseren Erfahrungen wird Rituximab bei MCD sehr gut vertragen, ein Tumorlyse-Syndrom wurde bislang nicht beobachtet. Hauptkomplikation der Rituximab scheint eine Reaktivierung eines Kaposi-Sarkoms zu sein, das wohl in etwa einem Drittel der Fälle auftritt. Auch auf infektiöse Komplikationen sollte insbesondere bei immunkompromittierten Patienten geachtet werden. Dazu zählen auch Erkrankungen wie die PML, die unter Rituximab auch bei HIV-negativen Patienten beobachtet wurde. Bei Rezidiven des MCD ist die erneute Gabe von Rituximab möglich. Bei drei Patienten, die nach 19-28 Monaten wieder Symptome eines MCD zeigten, wurden unter erneuter Rituximab-Monotherapie wieder mehrmonatige klinische Remissionen erzielt. In jedem Fall sollte die Krankenkasse vor einer Rituximab-Behandlung kontaktiert werden, da Rituximab bekanntlich sehr teuer und für den MCD nicht zugelassen ist.
ANDERE THERAPIEN
Neben Rituximab gibt es noch andere Ansätze, für die zumindest Fallberichte oder kleinere Fallserien existieren.
Valganciclovir: Wegen der engen Assoziation mit HHV-8 wurden einige antivirale Substanzen versucht, darunter vor allem Ganciclovir. Valganciclovir senkte in einer im Jahr 2008 veröffentlichen, doppelblind randomisierten Studie die HHV-8-Replikation deutlich und bietet sich daher für die MCD-Behandlung an. Eigene Beobachtungen und kleine Fallserien zeigten unter Valganciclovir Besserungen, teilweise in Kombination mit AZT-haltigen Regimen. Dagegen blieben Foscarnet oder Cidofovir in anderen Fällen ohne Benefit. Völlig unklar ist bislang, in welcher Dosis und wie lange Valganciclovir gegeben werden muss.
Chemotherapien: Gut verträgliche Substanzen wie Vincristin (2 mg i.v. als Bolus in 14-tägigen Abständen), Vinblastin oder orales Etoposid (täglich 50 mg) haben sich einigen Berichten zufolge und auch nach unserer Erfahrung als wirksam erwiesen. Auch eine CHOP-Chemotherapie kann durchaus helfen, scheint aber die Überlebenszeit nicht signifikant zu verlängern.
Immuntherapien: Ein immuntherapeutischer Ansatz ist Thalidomid, das die Zytokin-Dysregulation bzw. die inflammatorische Komponente unterdrücken soll und für dessen Wirkung es Fallbeispiele gibt. Eigene Erfahrungen bei inzwischen drei Patienten waren enttäuschend. Eine Besserung der Klinik wurde nicht beobachtet, stattdessen kam es zu schweren Komplikationen, darunter einer Lungenembolie. Möglicherweise eignet sich Thalidomid als prophylaktische Erhaltungstherapie, ein Schub lässt sich wohl kaum mildern. Für Interferon gibt es positive wie negative Fallbeispiele, ein Effekt ist fraglich. Steroide haben dagegen beim MCD dagegen sicher keinen Effekt.
Interleukin-6-Rezeptorblocker: Aus Japan gibt es Daten von einigen Dutzend HIV-negativen Patienten, die erfolgreich mit IL-6-Rezeptor-Antikörpern wie z.B. Tocilizumab behandelt wurden. Diese Antikörper befinden sich in Europa bei HIV-negativen Patienten mit Morbus Castleman bereits in klinischer Erprobung. Tocilizumab wird inzwischen unter dem Handelsnamen Actemra® für die Behandlung der Rheumatoiden Arthritis entwickelt. Valide Daten für den HIV-MCD liegen freilich bislang noch nicht vor.
Splenektomie: in schweren Fällen kann die Entfernung der Milz sinnvoll sein. Bei 40 Patienten lag das mediane Überleben der splenektomierten Patienten bei 28 vs. 12 Monaten. Einer US-Arbeitsgruppe zufolge besserte sich die Symptomatik bei 10/10 Patienten nach Splenektomie. Worauf dies zurückzuführen ist, weiß man allerdings bislang nicht. Spekuliert wird über eine Drosselung der IL-6-Produktion und darüber, dass durch die Splenektomie ein großes HHV- 8-infiziertes Zellreservoir entfernt wird.