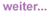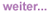Christian Hoffmann, Hamburg; Stefan Esser, Essen
Opportunistische Infektionen –Teil 7: Kaposi-Sarkom (KS)
Pathogenese, Epidemiologie
Der zelluläre Ursprung der typischen KS-Spindelzellen wird noch immer kontrovers diskutiert – neuere Untersuchungen lassen am ehesten lymphatische, endotheliale Zellen vermuten. Seit 1994 ist bekannt, dass das KS durch eine Infektion mit dem Humanen Herpesvirus 8 (HHV-8, von einigen Autoren auch als „Kaposi sarcoma-associated herpesvirus“ KSHV bezeichnet) mit verursacht wird. HHV-8 zählt zu den Gammaherpesviren und ähnelt dem Epstein-Barr-Virus. HHV-8 lässt sich fast obligat im Tumorgewebe nachweisen, die Höhe der HHV-8-Viruslast im Serum korreliert recht gut mit der KS-Progression. Übertragen wird HHV-8 wie andere Herpesviren überwiegend durch Speichel, aber auch sexuell, vertikal und wohl auch über Blut. Die Verbreitung von HHV-8 weltweit ist sehr unterschiedlich. Die Seroprävalenzen gesunder Blutspender reichen von 0,2 Prozent in Japan bis hin zu über 50% in manchen Regionen wie Zentralafrika, oder aber auch Italien und einigen anderen Mittelmeerländern. Obgleich eine entscheidende pathogenetische Rolle von HHV-8 in der der KS-Entstehung gesichert ist, sind die genauen Mechanismen nicht klar. Eine alleinige Infektion mit HHV-8 führt keinesfalls zwangsläufig zum KS. Interaktionen vor allem mit HIV, möglicherweise auch mit anderen Viren wie HHV-6 und HSV-1, veränderte Signaltransduktionsketten, eine erhöhte Produktion von Wachstumsfaktoren sowie Zytokindysregulationen spielen wahrscheinlich eine Rolle.
Unter den HIV-Patienten sind fast ausschließlich homosexuelle Männer betroffen; bei Frauen, Kindern oder auch Hämophilen ist das KS selten, kommt aber durchaus vor. Ein Immundefekt bzw. niedrige CD4-Zellen fördern Entstehung und Wachstum, sind aber keineswegs Bedingung. So ist das KS eine der wenigen Aids-definierenden Erkrankungen, die nicht selten auch schon bei gutem Immunstatus auftreten. Anders als für PCP, CMV-Retinitis oder Toxoplasmose sind 200 CD4-Zellen/µl für das KS kein Schwellenwert. Rund 29% aller Patienten, die in den USA in den Jahren 1996-2007 an KS-Studien teilnahmen, hatten über 300 CD4-Zellen/µl und eine Viruslast unter der Nachweisgrenze. In einer Studie korrelierte die Aktivierung der CD8-Zellen stärker mit der Progression als die CD4-Zellen. Auch gibt es, wenngleich selten, wenige Monate nach Einleitung einer ART zum Teil sehr aggressive Verläufe mit oft pulmonalem Befall im Rahmen eines Immunrekonstitutionssyndroms.

Abb. 1:
Pulmonaler KS-Befall
(mit frdl. Genehmigung Dr. Christoph Lange, Borstel)
Klinik
Das HIV-assoziierte KS befällt vor allem die Haut und die Schleimhäute. Oft sind jedoch auch Lymphknoten und innere Organe wie Gastrointestinaltrakt, Lunge oder Leber betroffen. Ein lymphatischer oder auch viszeraler Befall kommen mitunter auch isoliert vor, ohne dass die Haut beteiligt ist. Der Verlauf des KS ist sehr variabel und reicht von einzelnen, über Jahre stationären Läsionen bis zu ausgesprochen aggressiven, innerhalb weniger Wochen zum Tode führenden Verläufen. Auch ist der klinische Verlauf heute meist milder; insbesondere die früher schwer entstellenden und oft fatalen Manifestationen sind heute eine Seltenheit geworden. Probleme bereitet allerdings das KS im Rahmen eines Immunrekonstitutionssyndroms, das oft sehr rasch progredient ist und einer raschen chemotherapeutischen Behandlung bedarf.
Typischerweise zeigen sich KS-Läsionen anfangs als hell- bis lividrote, spindelförmige Flecken oder Knoten, die sich in Richtung der Hautspaltlinien anordnen. In der Mundschleimhaut ist oft der harte Gaumen betroffen. Prädilektionsstellen gibt es jedoch nicht – jede Lokalisation ist möglich. Insbesondere bei Befall sichtbarer Hautpartien ist das KS oft sehr stark stigmatisierend und für die Patienten eine erhebliche psychische Belastung.
Der weitere Verlauf kann erheblich variieren. Alles ist möglich: So können die Tumoren über Jahre auch ohne jede Therapie so gut wie unverändert bleiben, aber auch in wenigen Wochen sehr rasch wachsen und sich disseminiert ausbreiten. Rasches Wachstum geht mitunter mit Schmerzen und gelbgrünen Verfärbungen der Tumorumgebung (Einblutungen!) einher. Zentrale Nekrosen und Exulzerationen mit Blutungsneigung sind ebenso möglich wie zum Teil massive Ödeme, insbesondere an den Extremitäten, genital und im Gesicht. Abheilende KS blassen oft zunächst ab, verlieren an Größe und hinterlassen noch oft für Monate manchmal sogar lebenslang schmutzig-graubraune bis hellbraune Hyperpigmentierungen. Diese werden durch Hämosiderin-Ablagerungen, aber auch durch vermehrte Melanozytenstimulation infolge der Inflammation verursacht. Lymphödeme können besonders an den Unterschenkeln über Jahre persistieren.

Abb. 2:
Ulzeriertes Kaposi-Sarkom

Abb. 3:
Kaposi-Sarkom am harten Gaumen
Fotos: Dr.
Ramona Pauli, München
Diagnostik
Das kutane KS ist meist eine Blickdiagnose. Dennoch sollte großzügig und in allen nicht ganz eindeutigen Fällen eine Histologie (Exzision oder Inzision) entnommen werden. Nur so werden Differentialdiagnosen wie kutane Lymphome, ein Angiosarkom, ein Erythema elevatum et diutinum oder eine Bazilläre Angiomatose nicht übersehen. Auch eine Lues kann sich ähnlich darstellen. Histologisch finden sich die typischen spindelförmigen KS-Zellen, begleitet von lymphohistiozytären Infiltraten und schlitzförmigen, von Erythrozyten-Extravasaten umgebene Gefäßneubildungen.
Sofern die Diagnose eindeutig geklärt ist, sollte unbedingt der Ausbreitungsgrad des KS geklärt werden, und zwar mindestens durch:
- Komplette Inspektion (oraler und genitaler Schleimhäute! In den Mund gucken!)
- Sonografie des Abdomens
- Gastroduodenoskopie und Koloskopie (bei Befall der Schleimhäute obligat)
- Röntgen-Thorax (zum Ausschluss eines pulmonalen Befalls)
Laborbestimmungen helfen dagegen meist nicht weiter. Eine negative HHV-8-Serologie sollte Anlass sein, die Diagnose noch einmal zu hinterfragen. Eine HHV-8-PCR gibt lediglich Auskunft über die Virämie, etabliert als Routine-Parameter ist sie nicht. Der Immunstatus und die CD4-Zellen sollten natürlich bekannt sein.
Therapie
Bei bislang antiretroviral unbehandelten Patienten steht die Einleitung einer antiretroviralen Therapie an erster Stelle. Bei bereits antiretroviral behandelten Patienten ohne ausreichende HI-Virussuppression sollte die ART nach Möglichkeit optimiert werden. Mit Abfall der HI-Viruslast und einsetzender Immunrekonstitution stabilisieren sich die meisten mukokutanen KS oder heilen oft sogar ganz ab (bei allerdings oft zurückbleibenden postinflammatorischen Hyperpigmentierungen). In einer prospektiven Studie an 22 Patienten waren die KS-Läsionen nach 40 Monaten ART bei 18 Patienten komplett und bei 2 partiell abgeheilt – nur 2 waren progredient. Man kann also durchaus zunächst in vielen Fällen nur eine antiretrovirale Therapie geben und zuwarten.
Obwohl im Tierversuch ein direkter antiproliferativer Effekt für Proteasehemmer gezeigt wurde, gibt es keine „antiretrovirale Therapie der Wahl“. Auch NNRTI-haltige und andere ART-Kombinationen sind effektiv, die KS-Läsionen bilden sich unter diesen Kombinationen zurück. Entscheidend ist die Immunrekonstitution der Patienten. Therapiepausen sollten bei Patienten mit KS vermieden werden. KS-Rezidive auch nach langer kompletter Remission sind durchaus möglich. In der SMART-Studie war das KS während der Pausen eine der häufigsten AIDS-Erkrankungen, und zwar vor allem bei Patienten mit KS in der Vorgeschichte.
Eine alleinige ART reicht allerdings nicht bei
viszeralem Befall. In diesen Fällen, aber auch bei einem KS im Rahmen eines
IRIS ist immer eine systemische Therapie anzuraten. Auch bei raschem Progress
(vor allem im Rahmen eines IRIS), bei Persistenz trotz ART, aber auch bei
viszeralem, ausgedehntem mukokutanem KS-Befall oder bei Ödemen sollte ebenfalls
eine systemische Therapie eingeleitet werden. Abhängig von der Ausbreitung des
KS und des Immunstatus des Patienten bieten sich dabei mehrere
Behandlungsmethoden an, die im Folgenden besprochen werden sollen.
Medizingeschichte des KS
Im Jahre 1872 beschrieb der ungarische Hautarzt Moritz Kaposi fünf männliche Patienten mit „idiopathischen, multiplen, pigmentierten Sarkomen der Haut“. Diese inzwischen als „klassische“, nicht mit HIV assoziierte Variante des Kaposi-Sarkoms kommt vorwiegend bei älteren Männern aus Osteuropa bzw. dem Mittelmeerraum vor. Daneben gibt es in einigen Ländern südlich der Sahara noch das „endemische“ KS. Das „klassische“ KS ist relativ gutartig, befällt oft nur die Haut an den unteren Extremitäten und unterscheidet sich damit klinisch deutlich vom HIV-assoziierten KS, von dem im Folgenden die Rede sein soll.
Im Mai 1981, also über 100 Jahre nach Erstbeschreibung des KS, veröffentlichten die amerikanischen Centers of Disease Control, einen Bericht über fünf junge homosexuelle Männer mit Pneumocystis-Pneumonien und Zeichen einer Immunschwäche. Bereits im Juli, nur wenige Wochen später, wurden bei der gleichen Risikogruppe die ersten 26 KS-Fälle in New York und Kalifornien beschrieben, darunter ebenfalls einige Patienten mit PCP. So wurde durch die unerwartete und ansonsten extrem seltene Koinzidenz von PCP und KS deutlich, dass es sich um ein neues Krankheitsbild handelte: AIDS. Bis heute, inzwischen fast dreißig Jahre später, ist das KS der häufigste AIDS-definierende Tumor bei HIV-Patienten geblieben. Aufgrund seiner engen Assoziation zu HHV-8 wird er in dieser Serie der Opportunistischen Infektionen besprochen.
Liposomales Doxorubicin
Das pegylierte, liposomale Anthrazyklin Doxorubicin in einer Dosierung von 20 mg/m² Körperoberfläche ist unter den KS-Chemotherapien Mittel der Wahl. In bis zu 80% der Fälle wird eine Remission erreicht. Die Infusionen über 30-60 Minuten alle 2-3 Wochen sind ambulant machbar und meist gut verträglich. Eine antiemetische Therapie ist nicht erforderlich, eine Alopezie tritt üblicherweise nicht auf. Auf die korrekte Lage des intravenösen Zugangs ist unbedingt zu achten, da Doxorubicin komplikationsreiche Paravasate verursachen kann. Behandelt wird mindestens bis zur deutlichen partiellen Remission. Diese besteht, wenn die Tumoren ins Hautniveau abgeflacht sind und ein Farbwechsel von livide nach bräunlich und heller erkennbar ist. Die partielle Remission wird meist nach 6-8 Zyklen erreicht. Rezidive treten mit gleichzeitiger ART selten und dann vor allem im ersten Jahr auf. Wenn es längere Zeit nach dem Ende der Behandlung zum Rezidiv kommt, ist ein neuer Behandlungsversuch mit liposomalem Doxorubicin oft durchaus erneut erfolgreich. Ebenso hilft in Einzelfällen und bei ansonsten guter Verträglichkeit eine Therapieintensivierung mit Verkürzung der Zyklen und/oder einer Erhöhung der Dosis. Zu beachten sind die dabei jedoch die Myelotoxizität und die Kardiotoxizität. Obgleich letztere selten ist und erst oberhalb kumulativer Dosen von 450 mg vorkommt, sind transthorakale Echokardiographien (Auswurfleistung?) zu Therapiebeginn und Kontrollen nach jeweils 6 Zyklen, vor allem, wenn die Therapie ausgedehnt wird, dringend zu empfehlen. Eine weitere Nebenwirkung von liposomalem Doxorubicin ist die „palmoplantare Erythrodysästhesie“, die sich an Händen und Füßen als schmerzhafte makulöse Erytheme bemerkbar macht. Angesichts der potentiell immunsuppressiven Wirkung von Doxorubicin sollten alle Patienten mindestens für die Dauer der Therapie eine Cotrimoxazol-Prophylaxe erhalten, auch wenn die CD4-Zellen über 200 Zellen/µl liegen.
Auch das Taxan Paclitaxel ist beim KS effektiv. Es ist allerdings deutlich stärker myelotoxisch als liposomales Doxorubicin und führt zudem fast immer zur Alopezie, oft schon nach der ersten Gabe. Die Patienten sollten darüber unbedingt informiert werden. Paclitaxel sollte deshalb nur eingesetzt werden, wenn das KS unter Anthrazyklintherapie progredient ist oder rezidiviert. Neben Palcitaxel scheint auch Docetaxel unkontrollierten Studien zufolge wirksam zu sein. Bei den Taxanen müssen Interaktionen mit der ART beachtet werden. Ebenfalls als Rezidiv-Behandlung (nach Anthrazyklin- oder Paclitaxeltherapie) geeignet sind orales Etoposid oder Irinotecan.
Interferon
Mit Interferon (IFN) werden gute Remissionsraten erreicht, die allerdings wohl etwas niedriger sind als unter liposomalem Doxorubicin. Der Wirkmechanismus ist von Interferon auf das KS-Wachstum nicht geklärt. Neben der immunmodulierenden Wirkung induzieren Interferone aber wohl in KS-Zellen die Apoptose und hemmen die Angiogenese. Die Effektivität hängt deutlich ab vom Immunstatus. Oberhalb 400 CD4-Zellen/µl liegen die Remissionsraten bei über 45%, unterhalb 200 CD4-Zellen/µl bei nur 7%. Bei niedrigen CD4-Zellen verursacht Interferon nur Nebenwirkungen und sollte daher vermieden werden. Meist werden niedrige, subkutane Tagesdosen von 3-6 Mio. IE Interferon-a 2b verwendet, das im Gegensatz zu IFN- a 2a eine Zulassung für das KS hat.
Nach einer Anleitung durch medizinisches Personal kann der Patient die täglichen, subkutanen Injektionen selbst vornehmen. Mit einsetzender Remission (Stopp des Tumorwachstums, Abflachen der Tumoren, Verlust der lividroten Färbung, Wechsel in bräunliche, hellere Farbtöne) kann die Gabe auf dreimal/Woche reduziert werden. Pegylierte Interferone (wöchentliche Gabe möglich, insgesamt auch verträglicher als klassische Interferone) sind für das KS bislang nicht zugelassen, die optimale Dosis ist nicht bekannt. Sie sind aber wahrscheinlich mindestens genauso gut wirksam, wie erste Fallberichte bei AIDS-KS, aber auch beim klassischen KS vermuten lassen.
Neben der regelmäßigen Inspektion des Lokalbefundes an Haut und Schleimhäuten gehören Staging-Untersuchungen zur Therapiekontrolle. Diese sollten sich an der ini-tialen Lokalisation orientieren.
Andere Therapien
| Therapie | Dosierung | Kommentar |
|---|---|---|
| Liposomales Doxorubicin (Caelyx®) | 20 mg/m2 i.v. alle 2 Wochen | Therapie der Wahl, cave Myelo- und Kardiotoxizität, palmoplantare Erythrodysästhesie |
| Interferon-a 2a (Roferon®) | 3-6 x106 I.E. s.c. oder i.m. 3x / Woche, (Dosis je nach Verträg- lichkeit steigern) | Zum Teil erhebliche Nebenwirkungen, wohl etwas schwächer als Doxorubicin. Nur bei CD4-Zellen >200/µl und begrenztem Befall |
| Pegyliertes Interferon-a 2b* (PegIntron®) | 50 µg s.c 1 x / Woche. | Wie IFN-a (2a, b), aber besser verträglich, wenig Daten beim AIDS-KS! Vorsicht: Off Lable-Use |
| Paclitaxel (Taxol®) | 100 mg/m² i.v. alle 2 Wochen oder 135 mg/m2 i.v. alle 3 Wochen | Reservemittel Cave Neutropenie, periphere Neuropathie, Allergien, Alopezie Off Lable-Use! Vorsicht ART-Interaktionen |
Tab. 1: KS-Therapien, wenn die ART und Lokaltherapien nicht reichen
Lokaltherapien des KS sind verträglich und meist kostengünstig. Gerade einzelne ästhetisch oder funktionell störende Läsionen können mit einer Lokaltherapie schnell und effektiv behandelt werden. Vieles ist möglich: Einfache Camouflage, lokale Kompressionstherapie, aber auch Kryochirurgie, die rein intraläsionale Behandlung mit Vinca-Alkaloiden, Exzisionen, Bleomycin oder Interferon, oder auch Imiquimod-Creme.
Das KS ist auch strahlensensibel. Bei oberflächlichen makulösen oder plaqueförmigen Tumoren reichen Röntgenweichstrahlen in Einzeldosen von 4-5 Gy (Gesamtdosis 20-30 Gy, Fraktionierung 3x/Woche). Bei großflächigen KS mit Ödemen sollte dagegen mit schnellen Elektronen (5x 2 Gy pro Woche) bis zu einer Gesamtzielvolumendosis von 40 Gy bestrahlt werden.
Da das KS eine multilokuläre Systemerkrankung ist, macht eine Operation meist wenig Sinn. Wenn überhaupt, sollten nur kleine, kosmetisch auffällige Tumoren operativ beseitigt werden. Allerdings ist dabei mit Narbenrezidiven zu rechnen.
Bei KS-bedingten Lymphödemen, vor allem an den Extremitäten, sollten, sofern keine Kontraindikationen (schwere Herz- und/oder Niereninsuffizienz) bestehen, frühzeitig mechanische Lymphdrainagen und ggf. eine angepasste Kompressionstherapie erfolgen. Diese fördern die Rekanalisierung der geschädigten Lymphgefäße und verhindern Komplikationen wie chronische Ulzera.
Lokaltherapien haben im ART-Zeitalter allerdings deutlich an Bedeutung verloren; sie sind heute nur noch selten notwendig.
Neue Therapieansätze
Aufgrund der KS-Pathogenese wurden und werden immer wieder neue Therapien des KS vorgeschlagen, darunter Virusstatika, aber auch Zytokine und Angiogenese-Hemmer. Einige sollen im Folgenden stichwortartig erwähnt werden.
- Valganciclovir – senkt die HHV-8-Viruslast, wie unlängst eine randomisierte Studie zeigte. Valganciclovir ist damit ein sehr spannender Ansatz, zumal besser verträglich und praktikabel als Foscarnet, das in der 90ern mal im Gespräch war. Klinische Studien zum KS sind jedoch noch nicht veröffentlicht. Da HHV-8 an der frühen Tumorgenese beteiligt ist, bezweifeln einige Experten, ob überhaupt eine Wirkung bei einem bereits manifesten KS vorhanden ist.
- Interleukin-12 - zeigte hohe Ansprechraten in einer Phase-II-Studie, allerdings kombiniert mit liposomalem Doxorubicin. Randomisierte Studien fehlen.
- Sirolimus (und Everolimus) – neue Immunsuppressiva aus der Transplantationsmedizin. Wahrscheinlich wird die Tumor-Angiogenese durch eine verminderte Produktion vaskulärer endothelialer Wachstumsfaktoren gehemmt. In unkontrollierten Studien gute KS-Ansprechraten bei HIV-negativen Nierentransplantierten.
- Imatinib – bei Leukämien unverzichtbarer Tyrosinkinase-Hemmer, der wohl auch beim KS relevante Wachstums-faktoren (PDGF) hemmt. Partielle Remissionen bei 5/10 AIDS-KS-Patienten, auch histologisch.
- Sorafenib – neuer Tyrosinkinasehemmer beim Nierenzellkarzinom. Fallberichte zum KS. Aktuell laufen Phase-1-Studien.
- Retinoide (All-trans-Retinoinsäure) hemmen die KS-Proliferation – als topisch appliziertes Gel, als orale Kapseln oder auch intravenös liposomal modifiziert wurden einige Studien durchgeführt – die Response-Raten waren aber wohl nicht überzeugend, dass sich dieser Ansatz letztlich nicht durchgesetzt hat.
Literatur beim Verfasser