Nils Von Hentig, Frankfurt
HIV/HCV Koinfektion Pharmakologie der neuen Direct Acting Antivirals
Ansätze neuer Wirkstoffe
HC-Viren sind membranumhüllte, einzelsträngige RNA-Viren der Familie der Flaviviren.
Sie werden in sieben Genotypen unterteilt, die ihrerseits aus über 50 Subtypen bestehen. In Analogie zu anderen Flaviviren gelangt das HCV wahrscheinlich durch Clathrin-vermittelte Endozytose in die Zielzelle, wo es zur Fusion der Endosomenmembran mit der Virushülle und dadurch zur Freisetzung des Coreprotein-RNA-Komplexes in das Zytosol kommt. Auf die Freisetzung des viralen Genoms folgt die Synthese der Virusproteine an Membranen des endoplasmatischen Retikulums (ER).
Das als mRNA lesbare HCV-Genom gelangt zunächst direkt zu den Ribosomen des rauhen ER, wo eine erste Synthese von Virusproteinen stattfindet. Dieser erste Schritt ist notwendig, da das Virus zuerst einige Moleküle der viralen RNA-abhängigen RNA-Polymerase NS5B benötigt und dieses Enzym in der Zelle nicht vorhanden ist.
Um die Menge an verfügbarer viraler mRNA zu erhöhen, folgt als weiterer Schritt die Vermehrung der viralen RNA. Dazu synthetisiert die NS5B-Polymerase einen Negativstrang der viralen (+)RNA als Vorlage für die folgende Synthese weiterer Plusstränge. Als Initiationssignal für die NS5B-Polymerase dienen wahrscheinlich die spezifisch gefalteten Abschnitte in den beiden nichtcodierenden Regionen der viralen RNA. Es gibt Hinweise darauf, dass sich beide Enden des RNA-Genoms über die Bindung an zelluläre Proteine zu einem Ring schließen und dadurch ein Replikationskomplex entsteht. In HCV-replizierenden Zellkulturen konnten im Elektronenmikroskop auffällige Membranstrukturen gefunden werden, sogenannte membranous webs, die als HCV-Replikationskomplexe angesehen werden.
Nach Synthese der viralen mRNA beginnt im nächsten Schritt die Translation der viralen Proteine. Zur Initiation der Translation an den Ribosomen nutzen eukaryote mRNAs in der Regel eine Modifizierung des 5’-Endes, das sogenannte Cap. Das HCV besitzt durch die Faltung der 5‘-NCR eine besondere Struktur zur CAP-unabhängigen Initiation, die Internal ribosomal entry site (IRES). Dadurch benötigt die HCV-RNA keine zellulären Faktoren zur Bindung an die Ribosomen und zum Start der Proteinsynthese. Noch während der Translation werden die Strukturproteine von zellulären Proteasen vom entstehenden Polyproteinstrang abgeschnitten; die Hüllproteine E1 und E2 werden direkt in das ER synthetisiert, lagern sich in die Membran ein und die Glykolysierung wird im Golgi-Apparat fortgesetzt. Das Core-Protein wandert zuerst an sogenannte Lipid droplets. Diese liegen häufig in der Nähe der ER-Membran. An der Kontaktstellen zwischen Lipid droplets, HCV Replikationskomplexen und der ER-Membran findet dann wahrscheinlich die Assemblierung und das Budding ins ER statt.
Die neuen Virionen verlassen nun durch zelluläre Sekretion über den Golgi-Apparat die Zelle.
An einigen dieser Replikationsschritte setzen die neuen HCV-Medikamente an.
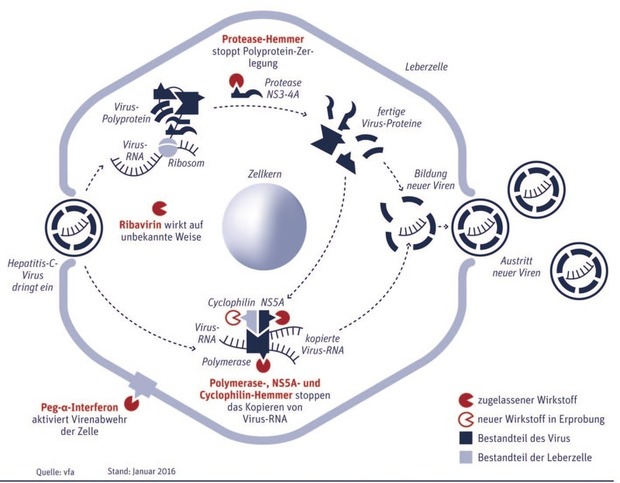
Abb. 1 Wirkprinzipien von Hepatitis-C-Medikamenten
Proteasehemmer (NS3/NS4A-Hemmer)
Das HCV-Virusgenom besteht – wie bereits beschrieben – aus einer einzelsträngigen RNA, welche nach dem Eintritt in die Wirtszelle direkt zur Translation, also zur Synthese der viralen Proteine genutzt werden kann. Diese werden zunächst in Form eines Vorläufer-Polyproteins synthetisiert: Dieses besteht aus mindestens 10 Funktionseinheiten, die mittels zellulärer und viraler Proteasen aus dem Vorläufer-Polyprotein geschnitten werden. Eine der viralen Proteasen liegt im Nicht-strukturprotein (NS3), das für seine Funktionalität einen viralen Kofaktor, das NS4A benötigt. Eine Hemmung diese Enzyme blockiert also die Herstellung funktionaler Virusproteine und damit die gesamte HCV Genomvermehrung.
Polymerasehemmer (NS5B-Hemmer)
Virusproteine allein formen aber noch kein vollständiges Virus. Dafür ist auch neues Viren-Erbgut nötig. Das wird in der Zelle mit der Hilfe zweier weiterer Proteine, die aus dem großen Virusprotein herausgeschnitten wurden, hergestellt: NS5A und die RNA-Polymerase (synonym: NS5B). Die RNA-Polymerase und NS5A synthetisieren zusammen mit dem zelleigenen Protein Cyclophilin B Kopien der Viren-RNA – An allen drei Proteinen lässt sich medikamentös ansetzen.
NS5B ist eine „klassische“ Polymerase, die aus 3 Domänen besteht, die RNA-Synthese de novo (also ohne primer) initiiert und das Plusstrang RNA Genom in eine Minusstrangkopie umschreibt, die ebenfalls durch NS5B zur Synthese vieler neuer Kopien der Plusstrang RNA genutzt wird.
Bei den NS5B Inhibitoren sind zwei Wirkstoffgruppen zu unterscheiden. Zum einen sind dies die Nukleoside, die dem natürlichen Substrat sehr ähnlich sind, aber auf Grund einer chemischen Modifikation zumeist am 2’-OH Ende nach Einbau in die virale RNA im Rahmen der Replikation zum Abbruch der RNA-Synthese führen. Diese werden im Sprachgebrauch als Kettenterminatoren bezeichnet. Zum anderen werden die nicht-nukleosidischen Polymerasehemmer, welche direkt die virale Polymerase hemmen, beschrieben.
Nukleos(t)idische Polymerasehemmer
Sofosbuvir ist ein Prodrug und ein Nukleotid-Analogon. Erst die intrazelluläre zweifache Phosphorylierung sorgt für die antivirale Aktivität. Als Uridintriphosphat-Analogon wird Sofosbuvir anstelle von Uridin als falscher Baustein in die virale RNA eingebaut und sorgt für einen Kettenabbruch. Insofern ist die Bezeichnung NS5B-hemmer nicht ganz zutreffend. Die meisten Kettenterminatoren zur Behandlung von Virusinfektionen sind Nukleosidanaloga, denen eine funktionale 3’-OH-Gruppe fehlt (z.B. Tenofovir, Aciclovir oder Azidothymidin). Aufgrund der Ineffektivität der Umsetzung von Nukleosiden mit fehlender 3’-OH-Gruppe in das Monophosphat, wurden für die Hemmung der HCV-Polymerase Analoga mit Modifikationen an der 2’-OH-Gruppe entwickelt.
Nicht-nukleosidische Polymerasehemmer
Während die nukleosidischen Inhibitoren das aktive Zentrum der Polymerase hemmen, binden die nicht-nukleosidischen Inhibitoren an eine allosterische Bindungsstelle des Enzyms. Das bedingt eine Konformationsänderung der Polymerase, welche zu einer Herunterregulierung ihrer Aktivität führt. Dabei sind allerdings verschiedene Bindungsstellen potenzeille targets der nicht-nukelosidischen Polymerasehemmer. Als eine Folge davon und aufgrund verschiedener Hemmmechanismen bzw. Unterschiede in der inhibitorischen Potenz, weisen sie eine relative zu Nukleosiden niedrigere Resistenzbarriere auf. „Außerdem zeigen die Nicht-Nukleoside eine zum Teil sehr ausgeprägte Genotypspezifität, während Nukleoside zumeist gegen viele Genotypen wirken“.
NS5A-Hemmer
Die Effekte beruhen auf der Bindung an das virale Protein NS5A (non-structural protein 5A). Im Unterschied zu anderen antiviralen HCV-Medikamenten handelt es sich beim drug target nicht um ein Enzym, sondern um einen Membran-assoziierten Phospholipidkomplex, welcher bei der RNA-Replikation und bei der Zusammensetzung des Virus (Assemblierung) eine Rolle spielt. NS5A interagiert mit viralen und zellulären Faktoren und spielt sowohl bei der RNA-Vermehrung als auch der Produktion infektiöser HCV-Partikel eine wichtige Rolle. Der erste zugelassene NS5A-Hemmer war Daclatasvir, welches direkt an NS5A bindet und wahrscheinlich auch dessen Multimerisierung verhindert. Es wird angenommen, dass hierfür die hoch symmetrische Struktur des NS5A-Hemmers verantwortlich ist, auch wenn dies bisher noch nicht direkt nachgewiesen werden konnte. NS5A-Multimere sind wahrscheinlich sowohl an der Vermehrung des HCV-RNA-Genoms als auch an der Assemblierung des Virus beteiligt. Dieser doppelte Wirkmechanismus könnte ein Grund sein, warum NS5A Inhibitoren eine hohe antivirale Potenz besitzen. Modell-Berechnungen von Guedj et al. haben gezeigt, dass Daclatasvir mit sehr hoher Effektivität sowohl die Virusvermehrung als auch Assembly und Freisetzung hemmt.
MHIV/HCV-Koinfektion
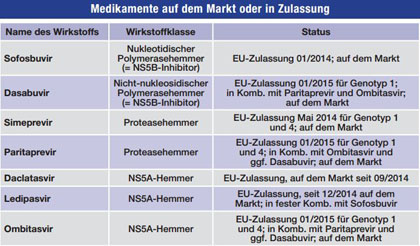
Tab. 1 Zur Behandlung der HCV-Infektion zugelassene neue Medikamente (Stand 23.12.2015)
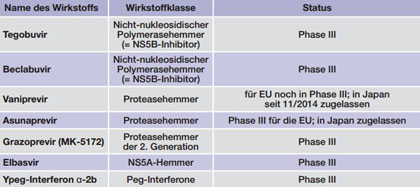
Tab. 2 Medikamente im Zulassungsverfahren oder Phase III (Stand 23.12.2015)
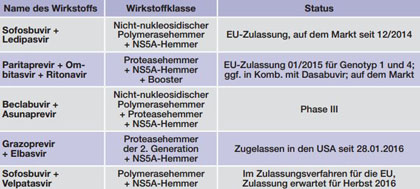
Tab. 3 Kombinationspräparate mit Zulassung oder in Phase III (Stand 23.12.2015
©Quelle: VFA
Die derzeit gültige deutsche Leitlinie zur Behandlung der Hepatitis C (Leitlinie der DGVS 2015) besagt, dass die „antivirale Therapie analog zu den Empfehlungen bei HCV monoinfizierten Patienten durchgeführt werden (Evidenzgrad IIb) sollte“. Grundlage dieser Empfehlung sind mehrere Studien, bei denen HIV/HCV-Koinfizierte Erwachsene genauso gut abschnitten wie HCV-Monoinfizierte. Von klinischer Relevanz bei HIV/HCV-Koinfizierten sind jedoch potentielle Wechselwirkung der antiretroviralen Substanzen mit DAAs. Mithilfe der Liverpooler Drug Interaction Database wurde bestimmt, in welchen Fällen die cART eine Kontraindikation zu verschiedenen DAAs darstellt bzw. potenzielle Interaktionen auftreten könnten. Dabei stellten die Autoren fest, dass die wenigsten Kontraindikationen/Interaktionen mit Sofosbuvir (0,2%/0%) auftreten, deutlich mehr mit Sofosbuvir/Ledipasvir (0,2%/67,6%), Daclatasvir (0%/ 49,4%), Ombitasvir/Ritonavir/Paritaprevir mit/ohne Dasabuvir (34,4%/ 52,2%) und mit Simeprevir (78,8%/0%). Die Substanzen können über verschiedene Wege interagieren.
Plasmaeiweißbindung
Die Plasmaeiweißbindung spielt eine Rolle bei der gleichzeitigen Gabe mit anderen, ebenfalls in hohem Maß an Plasmaeiweiß gebundene Arzneimittel (Vgl. auch Tabelle 4). Eine gegenseitige Verdrängung aus dieser Bindung bedingt eine höhere Konzentration freien Wirkstoffe im Plasma, so dass u.U. die Eliminationshalbwertszeit und die Wirkdauer eines Arzneistoffes verringert werden.
Leberstoffwechsel
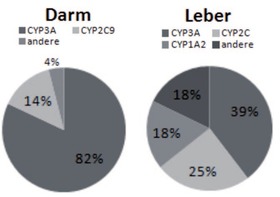
Abb. 2 Verteilung der CYP-Isoenzyme in Dünndarm und Leber
Cytochromoxidasen sind im humanen Arzneimittelstoffwechsel sowohl bei der Absorption im Darm als auch der hepatischen Metabolisierung involviert.
Etwa
60% aller Arzneimittel werden über hepatische Cytochromoxidasen
metabolisiert. Von diesen werden wiederum 40% über die
CYP3-Isoenzyme verstoffwechselt, 25% über CYP2C, 18% über CYP1A2
und 18% über andere. Die wichtigsten humanen Cytochromoxidasen sind
CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP2E1, and CYP3A4. Weitere wichtige
Stoffwechselenzyme sind Transferasen, wie z.B.
UDP-Glucuronyltransferasen (UGT) und N-Acetyltransferasen (NAT).
Aufgrund der Vielzahl und Komplexität der möglichen
Wechselwirkungen zwischen einzelnen DAAs und ARVs verweist der Autor
an dieser Stelle auf die jeweiligen Fachinformationen bzw. EMA-INN,
welche dieses Thema sehr detailliert in umfangreichen Tabellen
abarbeiten. Zusätzlich sei noch auf die Interaktions-Webseite der
Universität Liverpool hingewiesen, welche einen schnellen Überblick
für den ärztlichen Alltag bietet.
Zelluläre Transporter P-Glykoprotein (ABCB1)
Arzneistoffe müssen auf ihrem Weg in verschiedene Zielgewebe zelluläre Barrieren überwinden. Dies geschieht in vielen Fällen durch passive Diffusion, häufig aber auch aktiv mithilfe von Transportproteinen. Der bekannteste ist P-Glykoprotein (P-gp), welcher nach neuer Nomenklatur als ABCB1 Transporter (ATP-binding cassette B1) bezeichnet wird. Hierbei handelt es sich um einen Effluxtransporter, d.h. einen membranständigen ATP-abhängigen Transportmechanismus, welcher in der Lage ist, Toxine und auch als solche von der Zelle erkannte Arzneistoffe aus Zellen hinaus zu schleusen.
P-gp kann sowohl gehemmt als auch induziert werden, wobei Substrate von P-gp häufig auch Substrate von CYP3A4 sind (Tab. 4). Offenbar werden Expression und Aktivität beider Proteine zum Teil über die gleichen Mechanismen reguliert. Die Differenzierung der Effekte ist dadurch erschwert: Einige der Interaktionen, für die anfangs ein Angriff an CYP3A4 verantwortlich gemacht wurde, können auch durch Interferenzen mit P-gp oder durch beide Mechanismen verursacht sein.
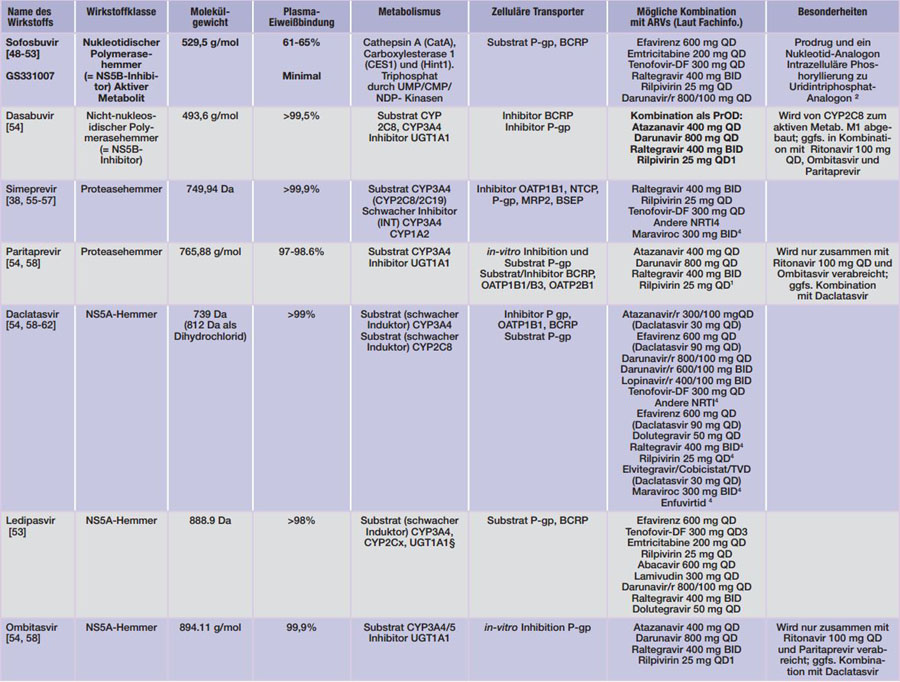
DA = Dalton; INT= Intestinal;HEP= Hepatisch.1 Nicht mit PI kombinieren, QTc-Zeit Verlängerung monitorieren. ²Uridin ist Bestandteil der HCV-RNA; 3
Kombination mit Atazanavir/RTV vermeiden, Kombination mit Efavirenz möglich, Kombination mit Darunavir/r QD bzw. Lopinavir/r 400/100 mg bzw. Elvitegravir/Cobicistat/Emtricitabin 150/150/200 mg nur unter Überwachung der Nierenfunktion.4 Kombination wurde nicht untersucht, es wird jedoch erwartet, dass keine klinisch signifikanten Interaktionen auftreten. §Widersprüchliche Aussagen in der EMA-INN ordnen Ledipasvir sowohl eine in vitro Induktion als auch Hemmung von CYP3A4 und UGT1A1 zu. INN S. 6.
Tab. 4 Pharmakologische Eigenschaften der derzeit zugelassenen HCV-Medikamente (Stand 23.12.2015)
Breast Cancer Resistance Protein (ABCG2)
Das humane Breast Cancer Resistance Protein (BCRP) ist ebenfalls Mitglied der ABC-Transporter-Familie (ABCG2). BCRP wurde ursprünglich in multiresistenten Brustkrebs-Zelllinien gefunden, welche gegenüber mehreren der Standard-Therapeutika, wie Mitoxantron, Topotecan und Methotrexat resistent waren. BCRP transportiert ebenfalls Nitrofurantoin, Prazosin, und andere Xenobiotika aus Zellen heraus. BCRP wird regelmäßig in Stammzellen gefunden und wird darüber hinaus in hohem Masse in Geweben des Dünndarms, von Leber, Plazenta und der Blut-Hirn-Schranke exprimiert.
Bile Salt Export Pump (ABCB11)
Das ABCB11 Gen codiert für einen ABC-Transporter mit dem Namen Bile Salt Export Pump (BSEP). BSEP ist ein Mitglied der MDR/TAP Unterfamilie der ABC Transporter und ist verantwortlich für den Transport von Cholat-Konjugaten aus den Hepatozyten in die Gallenflüssigkeit. Die Aktivität dieses Transporters ist die Hauptdeterminante für Gallensäurebildung und -fluss und genetische Polymorphismen des ABCB11 sind assoziiert mit der progressiven familiären intrahepatischen Cholestase (PFIC2) und in der Folge mit einem stark erhöhten Risiko für Leberzellkarzinome im jungen Lebensalter.
Multidrug Resistance-Associated Protein (MRP)
Auch die verschiedenen Typen des Multidrug Resistance-Associated Protein (MRP) gehören zu den ABC-Transportern und benötigen ATP. Die beiden MRP-Typen sind von Polymorphismen betroffen, die sich auf die Verfügbarkeit von Arzneimitteln auswirken und für Interaktionen prädisponieren können. MRP2–Inhibitoren sind beispielsweise Probenecid, Furosemid, Saquinavir, Ritonavir, Lamivudin, Emtricitabin, Abacavir, Efavirenz, Nevirapin, Cidofovir, Adefovir und Tenofovir oder auch Simeprevir. Das ist auch ein Grund für das Auftreten des Fanconi-Syndroms: Arzneimittel, welche MRP2 hemmen, bedingen eine Anhäufung von organischen Anionen in Zellen des renalen proximalen Tubulus, wodurch an dieser Stelle die mitochondriale DNA-Synthese gehemmt wird.
Organische Anionen- und Kationen-Transporter
Die sogenannten organic anion transport polypeptides (OATP) und organic cation transporters (OCT) kommen in vielen Geweben vor. Im Unterschied zu P-gp verbrauchen sie kein ATP. OATP transportieren beispielsweise Gallensäuren und anionische Arzneistoffe (Atorvastatin, Pravastatin, Benzylpenicillin, Digoxin, Fexofenadin, Levothyroxin). Es wird angenommen, dass einige Penicilline und Probenecid mit Methotrexat um die tubuläre Sekretion durch OATP3 konkurrieren und so dessen Plasmakonzentration erhöhen. Möglicherweise spielt eine Hemmung von OATP durch Gemfibrozil bei der Wechselwirkung mit Statinen (Cholesterol-Synthese-Hemmer) eine Rolle.
NTCP
Das Na+-taurocholate cotransporting polypeptide (NTCP) oder liver bile acid transporter (LBAT) ist ein Protein, dass in Menschen durch das SLC10A1-(solute carrier family 10 member 1) Gen codiert wird. Diese Na+-Gallensäure-Kotransporter sind membranständige Glykoproteine, welche am intrahepatischen Gallensäurekreislauf beteiligt sind. Zwei homologe Transporter reabsorbieren die Gallensäure, zum einen vom intestinalen Lumen, aus den Gallengängen und in der apicalen Niere (SLC10A2); der andere Transporter ist in der basolateralen Membran der Hepatozyten angesiedelt (SLC10A1). Darüber hinaus ist NTCP ein Oberflächenrezeptor, der für das Entry von Hepatitis B- und D-Viren in die Hepatozyten notwendig ist.